
どうもこんにちは、だいさくです。
みなさん見られましたでしょうか。
バイオジェン/エーザイのアデュカヌマブが米国で承認申請予定となってますね!
アデュカヌマブ 臨床第III相試験で得られた大規模データセットの新たな解析 結果に基づき、アルツハイマー病を対象とした新薬承認申請を予定
個人的にかなり疑問が残ったし、
僕はCNSの領域を自分で経験したことがあるわけではないので、
深いところまで予測することができませんが、
ちょっと思ったことなどを書いていきたいと思います。
なぜ?アデュカヌマブ が米国で申請!日本はどうなる?
まず、このアデュカヌマブに関してこれまでの流れを簡単に振り返ってみたいと思います。
アデュカヌマブはエーザイがスイスのNeurimmune社から導入し、
バイオジェンと共同で開発を進めていたBIIB037という開発コードのモノクローナル抗体です。
アデュカヌマブはAD(早期アルツハイマー病)になりにくい健常人のヒト組織から作り出し、
アルツハイマー型認知症の原因の一つだと仮定されていた、
アミロイドカスケード仮説に対する治療薬で、
認知症発症後のアミロイドβとタウの分解・排出促進を行い、
アミロイドβ量を低下させる作用があると仮定されております。
最終段階である第lll相まで進んでおりましたが、
残念ながら2019年3月にこのまま試験を継続しても、
主要評価項目をメットできない可能性が高いと判断され中止となりました。
詳しく知りたい方は当時書いたこちらの記事をご参照ください。
ネガティブデータが一転した今回の状況を整理してみる

今回、なぜ以前の試験中止となったネガティブデータが一転して、
ポジティブデータに変わり、承認申請の段階まで進んでいったのかを整理してみますが、
その要因を一言でいうと、
高用量群ではアデュカヌマブの臨床的効果が高いと判断されたからです。
少し長くなってしまいますが、結構重要なので是非ご一読ください。
そもそも今回のアデュカヌマブの第lll相試験のデザインは、
EMARGE試験(1,638人)、ENGAGE試験(1,647人)という、
2つの投与量の違う臨床試験を同時に進行させ、それらを合わせて有効性と安全性を評価する、
他施設共同、無作為化、二重盲検、並行群間比較、対照はプラセボという試験です。
なので、本来であれば、3,285人の患者さんに投与されその結果をみる予定でした。
2018年の10月頃に患者さんの登録は終了したとエーザイのプレスリリースがありますので、
患者の登録はすでに終わっております。
2019年3月に1,748人の被験者のデータを解析したところ、
主要評価項目の達成の可能性が低いと予測されました。
これはよく大規模臨床試験で用いられる無益性解析によって導き出されております。
無益性解析というのは、簡単に説明すると、
今後この試験を中断した方が良いか?を判断するための解析方法で、
よく抗がん剤の臨床試験でも用いられてる一般的な解析方法になります。
簡単にいうと試験続行を判断する中間解析のようなものになります。
無益性解析が間違っていたわけではない

今回なぜネガティブデータが一転してポジティブデータに早変わりしたかというと、
この無益性解析が間違っていたわけではありません。
バイオジェン及びエーザイの説明によると、
無益性解析は1,748人の被験者のデータセットを基にしていたが、
その後すでに登録され、新たに利用可能となった患者データ2,066人を含んで、
合計3,285人の被験者のデータを解析したところ、
無益性解析のデータとは異なる結果が得られたと述べられています。
特にEMERGE試験(1,638人)の方は、
事前に計画した主要評価項目において統計学的に優位な結果を示した(P=0.01)
しかし、ENGAGE試験については主要評価項目は達成できませんでしたが、
サブグループ解析においてはEMERGE試験を支持する結果であったとバイオジェンは考えている。
と述べられています。
なので、前述の通り、
このアデュカヌマブの試験は2つの投与量の違う試験が同時に進行している試験で、
恐らく、EMERGE試験が高用量の成績をみるための試験だったのだと思います。
さらにENGAGE試験でのサブセット解析というのは、
ENGAGE試験でも高用量の投与群が存在していて、
全体ではネガティブだが、高用量だけを抽出してみると、
EMERGE試験を支持する結果だったんだと考えられます。
その中で無益性解析の1,748人の解析ではわからなかったが、3,285人のデータを解析したら、
高用量の群では統計学的な有意差がでたという事になります。
またアデュカヌマブ低用量群及び高用量群において、プラセボと比較して、
アミロイドプラーク沈着の減少が確認された(P<0.001)
脳脊髄液中のタウレベルに関するバイオマーカーもその臨床所見を裏付ける結果であった。
さらに副作用の発現に関しても高用量群においても特段大きな問題がなかったため、
様々な有識者と協議を重ねた結果、FDAに申請する予定になったということになります。
なぜ無益性解析と結果が変わったのか?
なぜ無益性解析と今回の解析で結果が変わったのか?というところですが、
それは本来予測されていたものより、
アデュカヌマブ高用量の投与が拡大したことが原因であるとされています。
すなわち、解析対象被験者数の増加、高用量の平均投与期間が長くなったこと、
途中のプロトコル改訂によって高用量投与が多くの被験者に可能となった事、
などいくつかの複数の要因が関わったものと考えられているということです。
要は結果論ではないか!?

この試験の結果や詳細は10月のカンファレンスで説明される予定とのことですが、
この発表されているデータだけをみる限りただの結果論ではないかとどうしても思ってしまいます。
こんなことがまかり通って良いのでしょうか?
もともと予定していた、
EMERGE試験、ENGAGE試験2つの試験を合わせた元々予定されていた解析結果がまず重要で、
その結果がポジティブなのであれば、無益性解析の結果と乖離があったと判断されても、
特段変なことではないと思いますが、基本的にそういうことはありえないと思います。
(今の段階ではわからないですが)
でも今回それがありえたのは、高用量投与群だけ抽出してるからです。
こういうことって抗がん剤の試験でもよくあって、
全体的にはネガティブデータだったけど、
ある一つの群だけを抽出してレトロで見てみると有意差がついていたみたいなことは、
しょっちゅうあるわけです。
しかし、それでその患者群だけ承認申請されるなんて事は、
僕は癌の領域では見たことがありません。
なぜなら、その特定の患者群だけを対象に改めて前向きな試験を行うと、
大抵ネガティブデータになってしまうからです。
だから今回のアデュカヌマブのようなパターンでも、
設定を高用量にして改めて前向き試験を行うべきだと思います。
結局レトロはレトロなので、絶対に前向き試験を行わないとダメだと思います。
ポジティブ試験の中のレトロ解析で、
もっと効果のある患者群を断定するとかいう話ではないのです。
この試験の中で高用量群が何人いたのかとかもわからないですが、
ネガティブ試験の中のレトロ解析を用いて承認申請って、
CNSの世界ではありえる事なのでしょうか。
僕はCNSの領域を自分で担当したことがないのわからないのですが、
もしご存知の方がいらっしゃったらぜひ教えていただけませんでしょうか。
アデュカヌマブは日本ではどうなる?

では今回はFDAに申請となっておりますが、
日本では一体どうなるのでしょうか。
詳細はわかりませんが、この試験が走る前のバイオジェンの社長のインタビュー記事には、
日本人は150人登録されることが目標とされているので、
試験の対象として日本の医療機関も参加されており、
恐らく日本人の患者さんもこの試験に参加されていると思います。
なので、従来通りだと日本でも申請はされるし、
保険適応になるのではないかと考えられます。
高用量群では25%進行のリスクを軽減するとされているので(レトロだけど)、
期待値がかなり高いのは事実で、
アンメットニーズど真ん中なのは間違いないので、
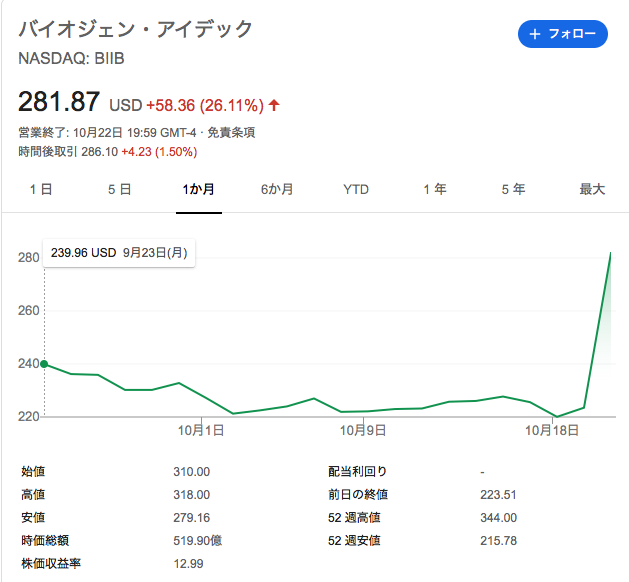
実際それを見越して、エーザイとバイオジェンの株はびっくりするぐらい爆上がりしました。

最後に
今回の結果はツイッター見ていても結構疑問を抱いてる方が多かったように思いますが、
皆さんはどう思われましたでしょうか。
今日本では、ご存知の通り企業主導試験が非常にやりづらい状況です。
アデュカヌマブが日本で上市されても、
それを裏付ける臨床試験を企業主導でやる事は戦略的にも100%ありえないと思います。
医師主導でやる事は考えられますが、
それを簡単に許す環境ではないし、
医療資源も余ってないと感じます。(CNSの世界をよく知りませんが)
このまま日本で承認されたら使われるだろうなぁってやっぱ思ってしまいます。
でも、結果はレトロだし、ただの結果論なのは間違いないので、
堂々と前向きに高用量でやったら良いと思うんですけどね。
これが標準療法がそれなりにある領域だったら、
こんな特別待遇はありえないっすからね。
ん〜医薬品業界というのは難しいですね。
ではまた!


コメント
こちらのサイトを知り、いろいろと過去にさかのぼって読ませていただきました。
(ちなみに業界のものではありますが、MR経験はありません。)
Aduについては、私もこれまでのよくある流れから、FDAから追加試験を要求されるであろうと考えていました。しかし、確認できた情報は、FDAのconsultationの結果、追加試験なしに秋口に申請されるだろうとのことでした。
サブ解析をもって、特定の患者群もしくは投与量群に対する評価を行うことは一般的なことだと思うので、こちらでの結果が、よほど説得力があるものだと想像しています。
尚、バイオジェンでは、これまでスピンラザの責任者をされていた方を既にCNSのヘッドに据えていますので、十分な臨戦態勢にはいっていると理解しています。
p.s.
株価チャートで、バイオジェン・アイデックとまだ表示されているのに、少し驚きました。
コメントありがとうございます。
アデュカヌマブは謎です。
何故こんなにFDAが優遇するのかもよくわかりません。
陰謀でしょうか(笑)
勉強不足で恐縮なのですが、アイデックという名前が表示されるのは何かあるのでしょうか??
数年前に、すでに社名をバイオジェンに変更しています。かつてサノフィAventisで後ろの名前が削除された様に、合併の力関係を示す典型例でした。参照された株価チャートの出展元のシステムで、まだ更新されていないのかなという感じだったので、コメントさせていただいた次第です。
ちなみに、AduもしくはBiogenを優遇するという感じではなさそうです。国内で仮に承認され、承認申請概要が公開されたら、どういう結果となったか見てみたいと個人的には思っています。でも、前のコメントで書いた通りで、通常の流れとは大きく異なっていたので、凄い結果になったのではと想像しています。
ありがとうございます!
申請概要、僕も注目したいと思います!!
確かに異例ということはそれなりに大きな理由がありそうですよね!